
立法基础与目标
医疗器械监督管理条例自2000年开始实施,旨在规范医疗器械的生产、流通和使用,保障人民群众的生命安全和身体健康。通过严格的法律法规,对医疗器械从研发到市场使用全程进行监管,以确保医疗器械品质高效、安全可靠。
注册审批制度
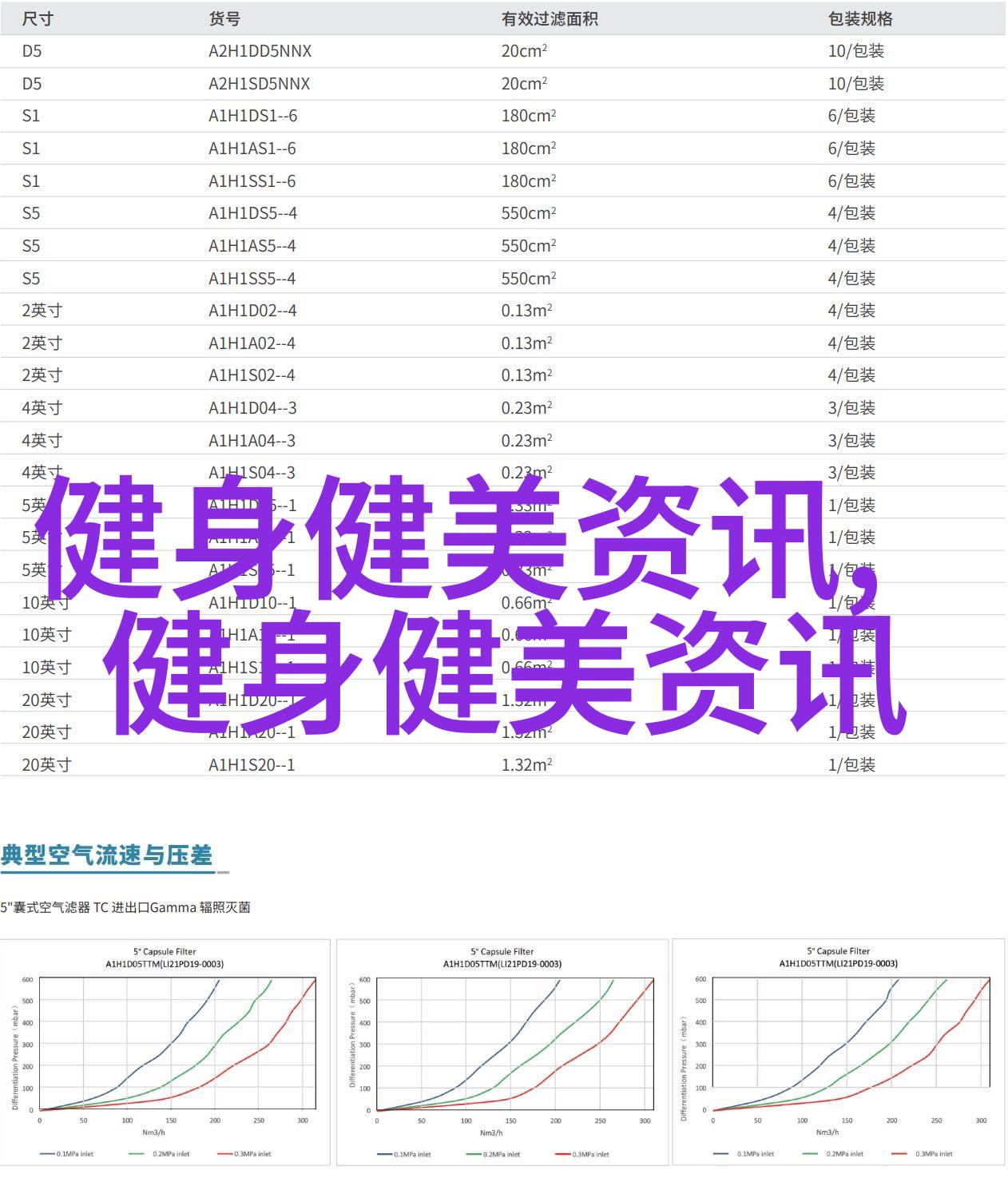
条例明确了医疗器械分类标准,将其分为三类:一类是需要临床试验验证后才能上市销售;二类是可以根据产品性能标准直接上市销售;三类是不需要特殊审批即可购买和使用。此外,还设立了预先评审、初步评审、终末评审等多个环节,对于不同类型的医疗器械实行不同的监管策略。
质量控制与追溯体系

为了提高产品质量,条例要求生产企业建立完善的质量管理体系,并对产品进行严格检验。同时,由于一次性医用非消毒棉签事件等事故频发,该条例还强调建立有效追溯系统,为不良反应及时跟踪并采取相应措施,是保护消费者权益的一个重要保证。
监督检查与处罚机制

针对违反规定的情况,该条例明确了相关部门应当加大对违法行为的查处力度,并提供了一系列处罚措施,如警告、罚款、责令改正或者暂停或吊销许可证等。在此基础上,还需不断加强执法人员培训,加大技术装备投入,以提升执法效能。
国际合作与交流

随着全球化进程不断深入,国内外医学技术水平日益接近,这也促使我国在国际范围内更加注重自身优势,同时学习借鉴其他国家在医疗器械领域成功经验。通过参加国际会议,与各国交流合作,不断丰富自己的法律知识库,为本国产业发展提供更多可能。